▪ EU-HTA in der Praxis: Seit Januar 2025 läuft die schrittweise Einführung der gemeinsamen europäischen Nutzenbewertung für Medikamente parallel zum Zulassungsverfahren. Im Mai 2026 startete in Deutschland das erste AMNOG-Verfahren auf dieser neuen Basis.
▪ Hoffnungsträger mRNA: Nach den Erfolgen in der Pandemie werden mRNA-Impfstoffe in klinischen Studien gezielt gegen Krebsarten wie Darm- und Hautkrebs getestet, um das Immunsystem auf Tumorzellen anzusetzen.
▪ Die Rolle der Daten: Für die Bewertung des tatsächlichen Zusatznutzens von Therapien gewinnen systematisch erhobene Routinedaten aus dem Klinikalltag massiv an Bedeutung.
▪ Wettbewerb im Pharmasektor: Effiziente Zulassungs- und Erstattungsverfahren entscheiden darüber, ob Europa im globalen Wettlauf um medizinische Innovationen gegen die USA und China bestehen kann.
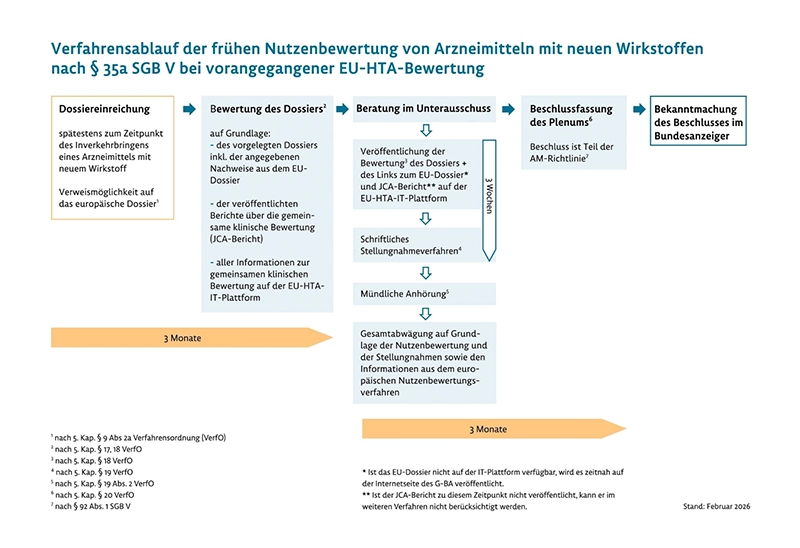
Seit Januar 2025 wird die europäische Nutzenbewertung (EU-HTA) schrittweise Teil des Marktzugangs neuer Arzneimittel. Das Ziel der EU-Verordnung 2021/2282 ist es, klinische Bewertungen auf europäischer Ebene gemeinsam zu erstellen, nationale Prozesse zu entlasten und Doppelarbeit zu vermeiden.
Im Jahr 2026 steht das System vor der ersten Reifeprüfung: Am 15. Mai 2026 startete das erste deutsche AMNOG-Verfahren auf Basis einer vorherigen europäischen Bewertung. Ein Beschluss des Gemeinsamen Bundesausschusses (G-BA) wird für November 2026 erwartet. Diese Neuerung betrifft in der aktuellen Phase vor allem Onkologika und neuartige Therapien (ATMPs). Bis 2030 soll das gemeinsame Verfahren auf alle zentral zugelassenen Arzneimittel ausgeweitet werden.
So informiert das IQWiG: „Gegenstand der ersten Gemeinsamen klinischen Bewertung (Joint Clinical Assessment, JCA) war das Krebsmedikament Tovorafenib zur Behandlung des pädiatrischen niedriggradig malignen Glioms. Für Deutschland hat das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) als Co-Assessor mitgewirkt. Gemeinsam mit dem National Center for Pharmacoeconomics aus Irland erstellte es den Bericht.“
Genau in diese erste Phase der EU-HTA-Regulierung fallen Krebsbehandlungen auf mRNA-Basis. Wirkstoffe dieser Art transportieren den genetischen Bauplan von Proteinen in die Zellen, um das körpereigene Immunsystem gezielt gegen Tumorzellen zu aktivieren. Diesen medizinischen Wendepunkt dokumentiert auch der Deutschlandfunk-Beitrag „Impfen mit mRNA: Erst Corona, jetzt der Krebs“, der den Weg der mRNA aus der Infektionsbiologie in die Onkologie nachzeichnet.
Klinische Studien untersuchen derzeit die Wirksamkeit vakziner Ansätze, die individuell auf die genetischen Veränderungen (Neoantigene) des jeweiligen Tumors abgestimmt sind. So laufen am St. Josef-Hospital der Ruhr-Universität Bochum Studien zu Dickdarm- und Bauchspeicheldrüsenkrebs. Am Universitätsklinikum Hamburg-Eppendorf wird dieser Ansatz zudem bei schwarzem Hautkrebs (Melanom) in Kombination mit Checkpoint-Inhibitoren getestet, um die körpereigene Immunantwort weiter zu verstärken.
Um den steigenden Bedarf an diesen personalisierten Therapien künftig decken zu können, kooperieren sieben Fraunhofer-Institute im Projekt „RNAuto“. Sie entwickelten eine automatisierte, digital gesteuerte Produktionsplattform, die mRNA-Wirkstoffe mittels digitaler Zwillinge und integrierter Sensorik skalierbar und kostengünstiger herstellen kann. Eine zeitgleiche EU-HTA-Nutzenbewertung könnte solche technologischen Entwicklungen schneller in die Versorgung bringen.
Ob eine innovative Therapie wie eine mRNA-Impfung von den Gesundheitssystemen erstattet wird, hängt elementar von der Generierung valider Daten ab. Für die Rückmeldung des nationalen Bewertungsumfangs (die sogenannte PICO-Fragestellung) ist in Deutschland der G-BA verantwortlich. Im Rahmen der gemeinsamen klinischen Bewertung (JCA) auf EU-Ebene werden die eingereichten klinischen Daten evaluiert, während die Entscheidung über den Zusatznutzen weiterhin bei den Mitgliedstaaten verbleibt.
Hierbei gewinnen neben klassischen klinischen Studien zunehmend routinemäßig erhobene Gesundheitsdaten („Routinely Collected Data“, RCD) an Bedeutung. Ein internationales Forschungskonsortium unter Beteiligung der LMU München und des Universitätsklinikums Bonn hat im Fachjournal The BMJ einen Leitfaden zur Nutzung solcher Routinedaten veröffentlicht. Große Patientenkollektive lassen sich so unter realen Versorgungsbedingungen abbilden, um die Qualität und Transparenz der evidenzbasierten Medizin zu stärken. Solche Daten sind für Institutionen wie das IQWiG wichtig, um den langfristigen Nutzen von Therapien im Behandlungsalltag zu überprüfen.
Für die pharmazeutische Industrie erwächst aus der Verknüpfung von EU-HTA, Studiendaten und Produktion ein spürbarer Handlungsdruck. Pharmazeutische Innovationen entstehen in einem globalen Wettbewerb; Investitionen fließen bevorzugt in Regionen mit verlässlichen und schnellen Zulassungs- und Erstattungsverfahren. Europa steht hierbei unter Druck durch die USA und China. Fragmentierte nationale Verfahren schwächen die Attraktivität des Standorts, weshalb eine effiziente Verzahnung der europäischen Prozesse gefordert wird.
Gleichzeitig stellen personalisierte Therapien wie mRNA-Krebsimpfstoffe, deren Jahrestherapiekosten pro Patient hoch ausfallen können (Schätzungen zufolge bei über 100.000 Euro), die Gesundheitssysteme vor finanzielle Fragen. Branchenverbände wie der BPI und Pharma Deutschland fordern daher angepasste Bewertungs- und Vergütungsmodelle, wie etwa Pay-for-Performance-Ansätze, um den Marktzugang für Onkologika zu sichern. Die deutsche Politik versucht, die Rahmenbedingungen über das Medizinforschungsgesetz (MFG) sowie den strukturierten „Pharmadialog“ zu steuern, welcher im Jahr 2026 in eine nationale Pharma- und Medizintechnikstrategie einfließen soll.
Deutschlandfunk-Beitrag zum Thema hier zum Nachhören (31:24).
Publikation: Sabine Hoffmann et al.: Using routinely collected data for research purposes: Challenges and mitigation strategies; The BMJ; DOI: https://doi.org/10.1136/bmj-2025-087812
IQWiG | EU-HTA-Verfahren: Erste Bewertung abgeschlossen

Comprix
Langzeit-Ranking
TOP 3
Zertifizierter Google Partner
seit 2017

Mitglied im GWA – Deutschlands
führende Agenturen